" L'étude de l'hypercholestérolémie familiale a prouvé que la compréhension d'une anomalie génétique rare pouvait éclairer un mécanisme fondamental de la biologie humaine pour le bénéfice de tous." (Cette phrase fait référence aux travaux fondateurs de Michael S. Brown et Joseph L. Goldstein, prix Nobel 1985, dont les recherches sur l'hypercholestérolémie familiale ont conduit à la découverte du récepteur des LDL).
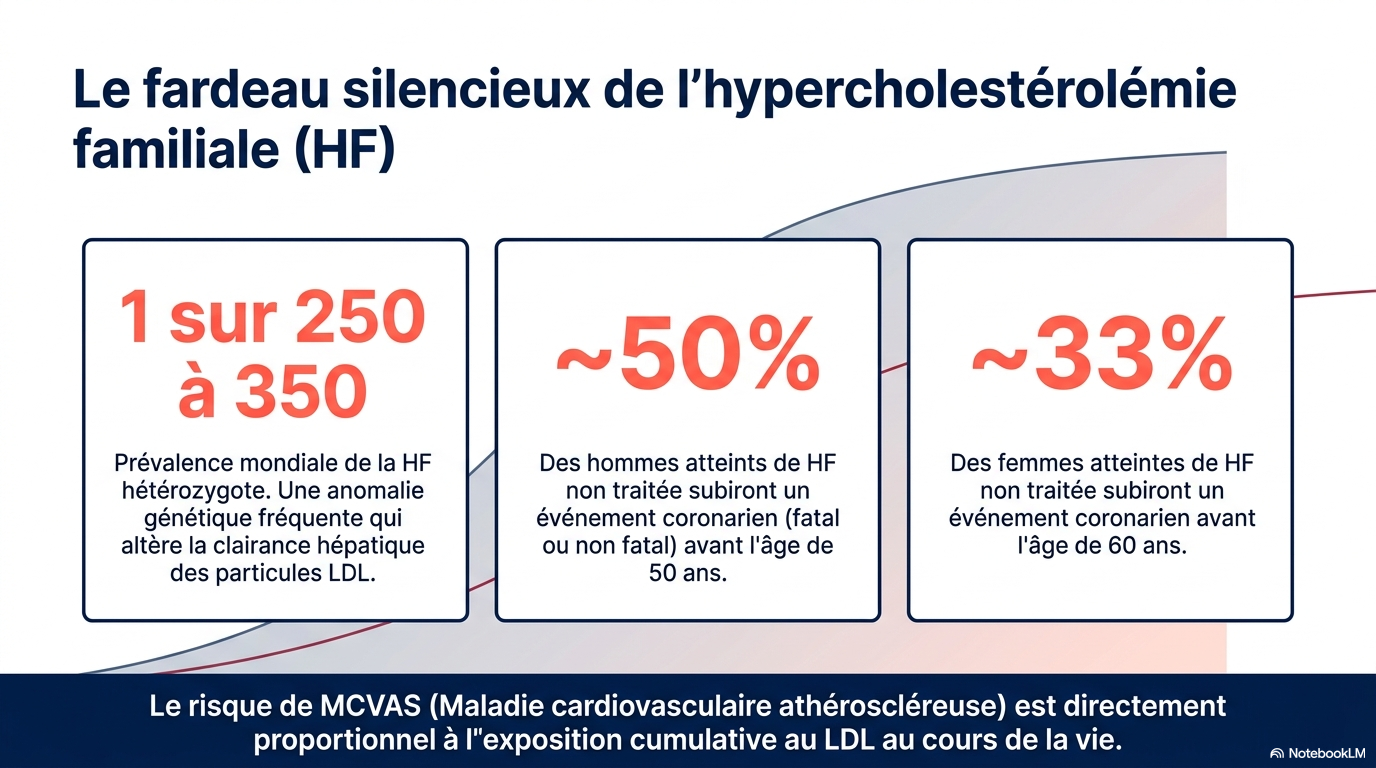
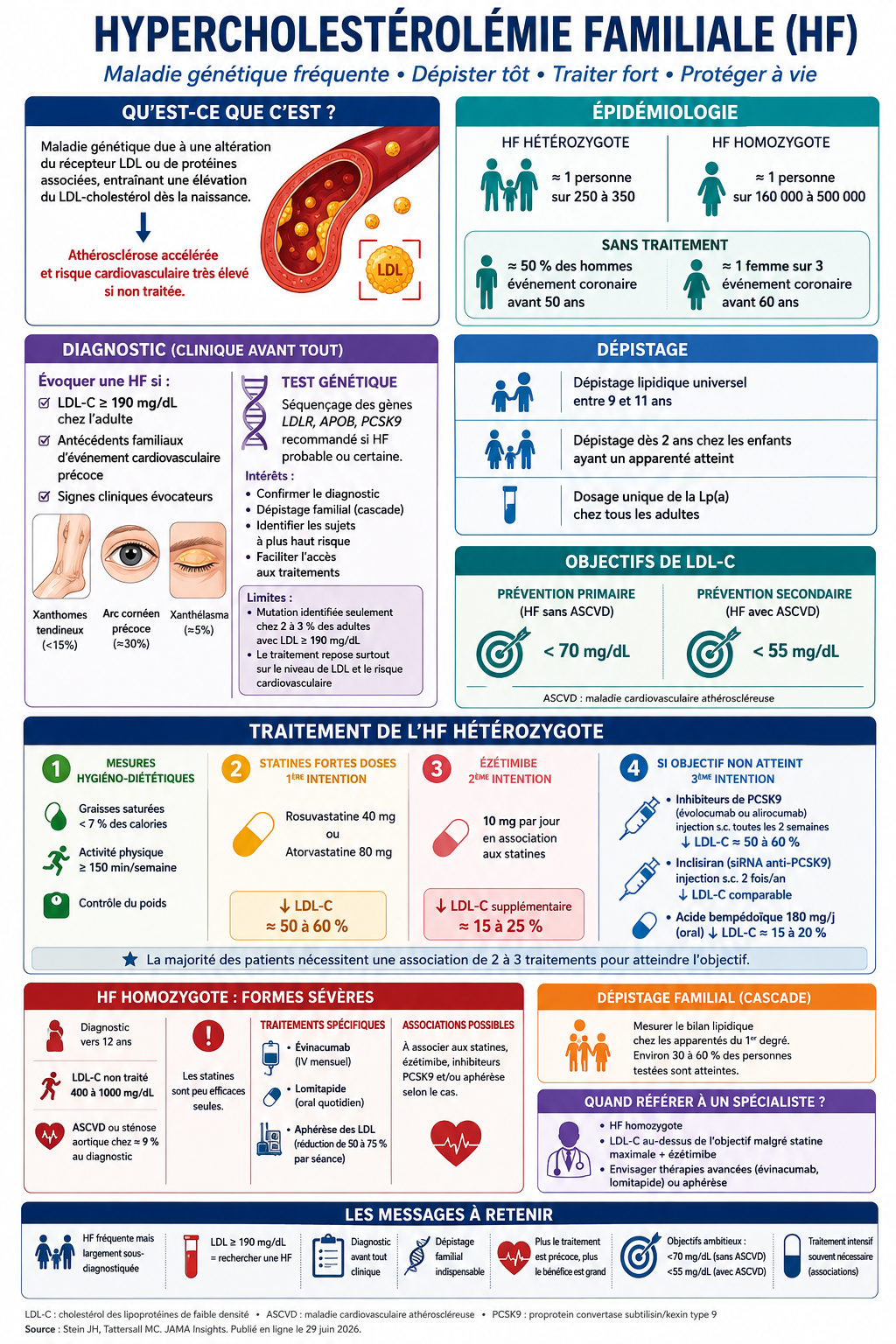
L’hypercholestérolémie familiale (HF) est un ensemble de maladies génétiques caractérisées par une altération de la clairance hépatique des lipoprotéines de basse densité (LDL), due à des variants pathogènes qui modifient la fonction des récepteurs LDL.élévation prolongée du cholestérol LDL (LDL-C) entraîne une athérosclérose accélérée et un risque accru de maladies cardiovasculaires athéroscléreuses (MCVA). À l’échelle mondiale, l’HF hétérozygote touche environ 1 personne sur 250 à 350, selon des études d’épidémiologie génétique portant sur les variants pathogènes des gènes associés à l’HF. L’ HF homozygote est rare, survenant chez environ 1 personne sur 160 000 à 500 000.Parmi les personnes atteintes d’HF hétérozygote non traitée, environ la moitié des hommes développent un événement coronarien fatal ou non fatal avant l’âge de 50 ans et environ un tiers des femmes avant l’âge de 60 ans.
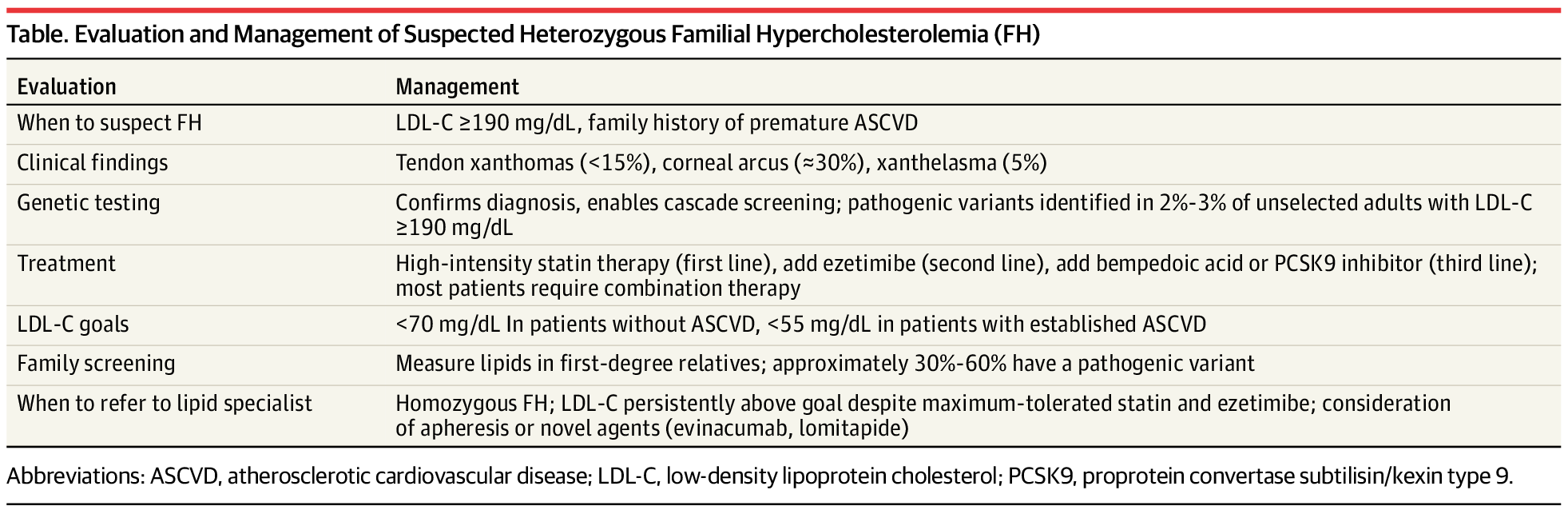
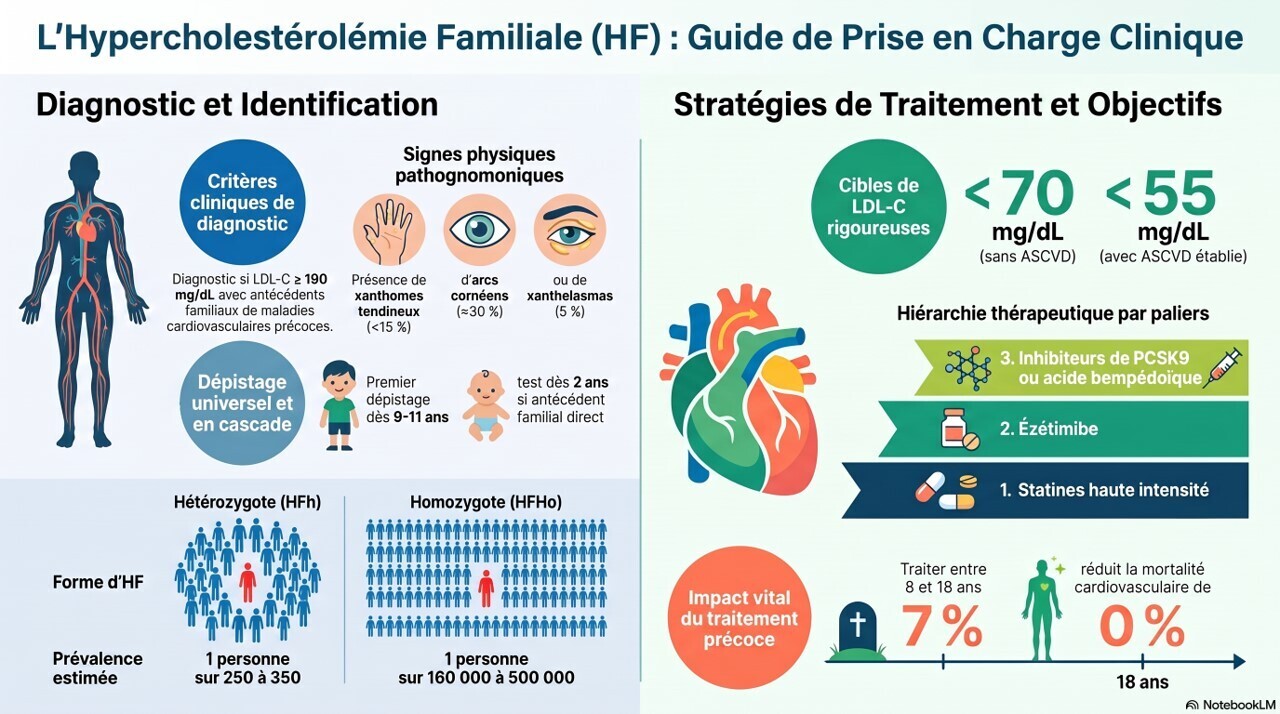
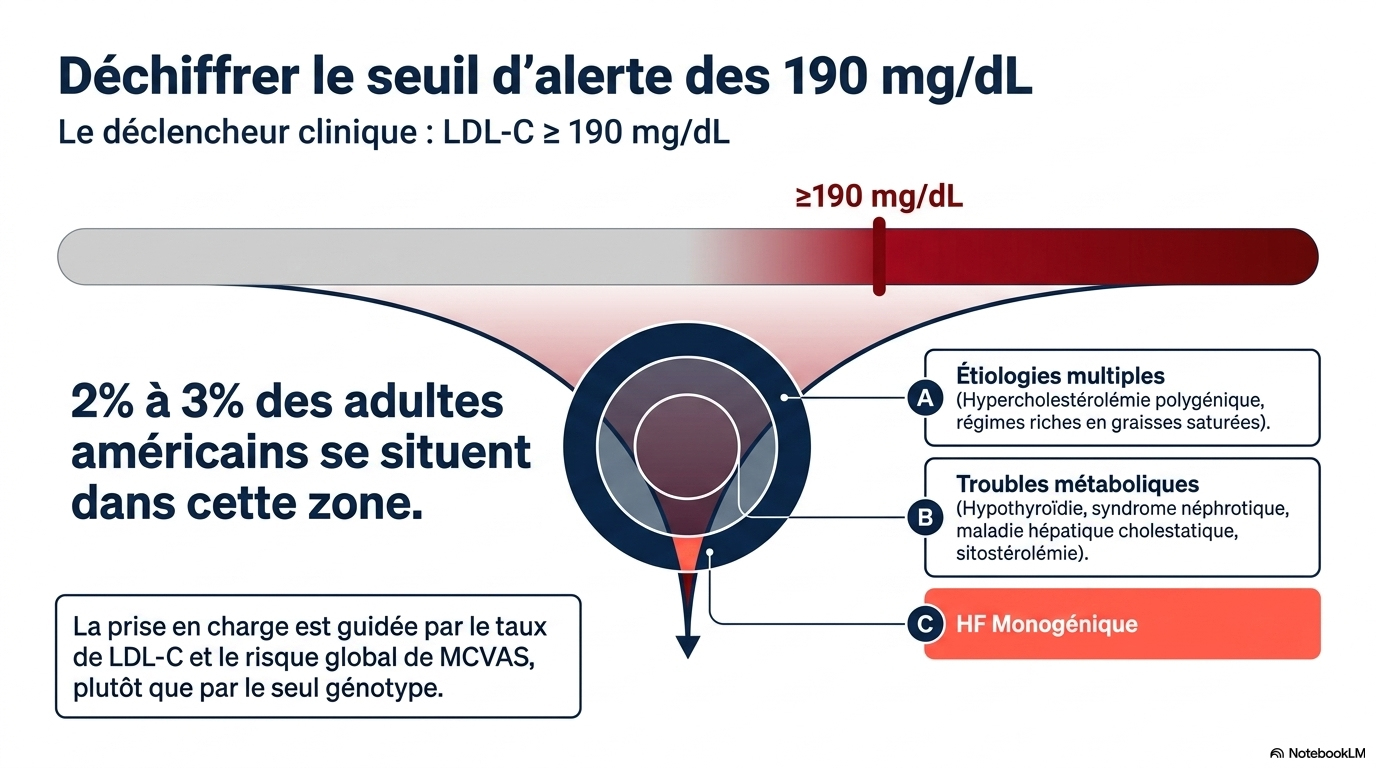
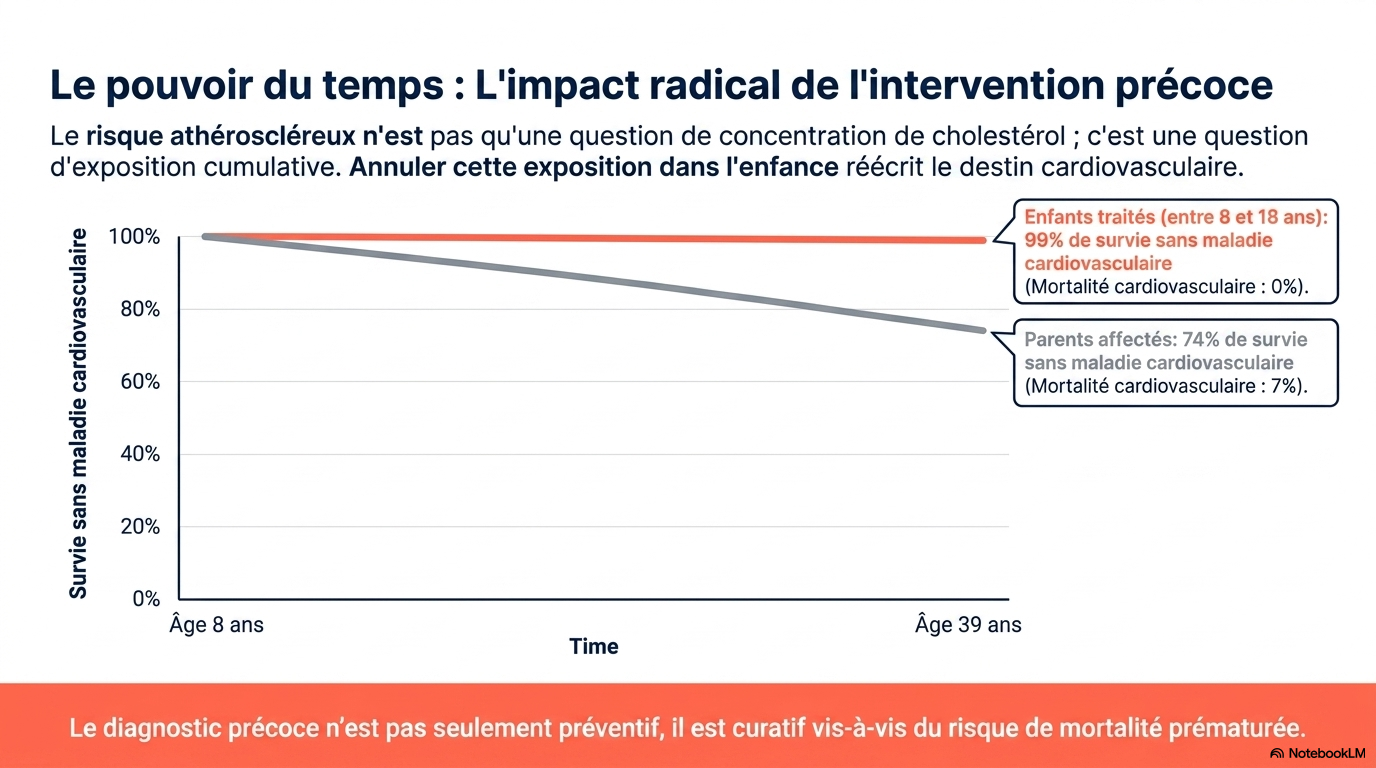
Le diagnostic d'hypercholestérolémie familiale (HF) repose sur un taux de LDL-C supérieur ou égal à 190 mg/dL et sur des antécédents personnels ou familiaux de maladie cardiovasculaire athéroscléreuse (MCVA) précoce. Il ne nécessite pas de confirmation génétique, bien que les tests génétiques permettent un dépistage en cascade chez les apparentés au premier degré. Le risque de MCVA étant lié à l'exposition cumulative au LDL-C dès le plus jeune âge, un traitement pharmacologique initié pendant l'enfance ou le début de l'âge adulte offre le meilleur bénéfice à vie. Le traitement de première intention est une statine à forte dose ; la plupart des patients nécessitent l'ajout d'ézétimibe et d'un inhibiteur de PCSK9 pour atteindre les objectifs de LDL-C : moins de 70 mg/dL chez les patients sans MCVA et moins de 55 mg/dL chez ceux présentant une MCVA avérée.



Résumé – Hypercholestérolémie Familiale (HF) / CHATGPT
D'après Stein JH & Tattersall MC. JAMA Insights, publié le 29 juin 2026.
Message clé
L'hypercholestérolémie familiale (HF) est l'une des maladies génétiques les plus fréquentes. Elle entraîne une élévation du LDL-cholestérol dès la naissance, responsable d'une athérosclérose accélérée et d'un risque cardiovasculaire très élevé si elle n'est pas diagnostiquée et traitée précocement.
Épidémiologie
- HF hétérozygote : environ 1 personne sur 250 à 350.
- HF homozygote : très rare (1/160 000 à 1/500 000).
- Sans traitement :
- près de 50 % des hommes présentent un événement coronaire avant 50 ans ;
- environ 1 femme sur 3 avant 60 ans.
Diagnostic
Le diagnostic est avant tout clinique.
Il faut évoquer une HF devant :
- LDL-C ≥ 190 mg/dL (4,9 mmol/L) chez l'adulte ;
- antécédents familiaux d'événement cardiovasculaire précoce ;
- ou présence de signes évocateurs :
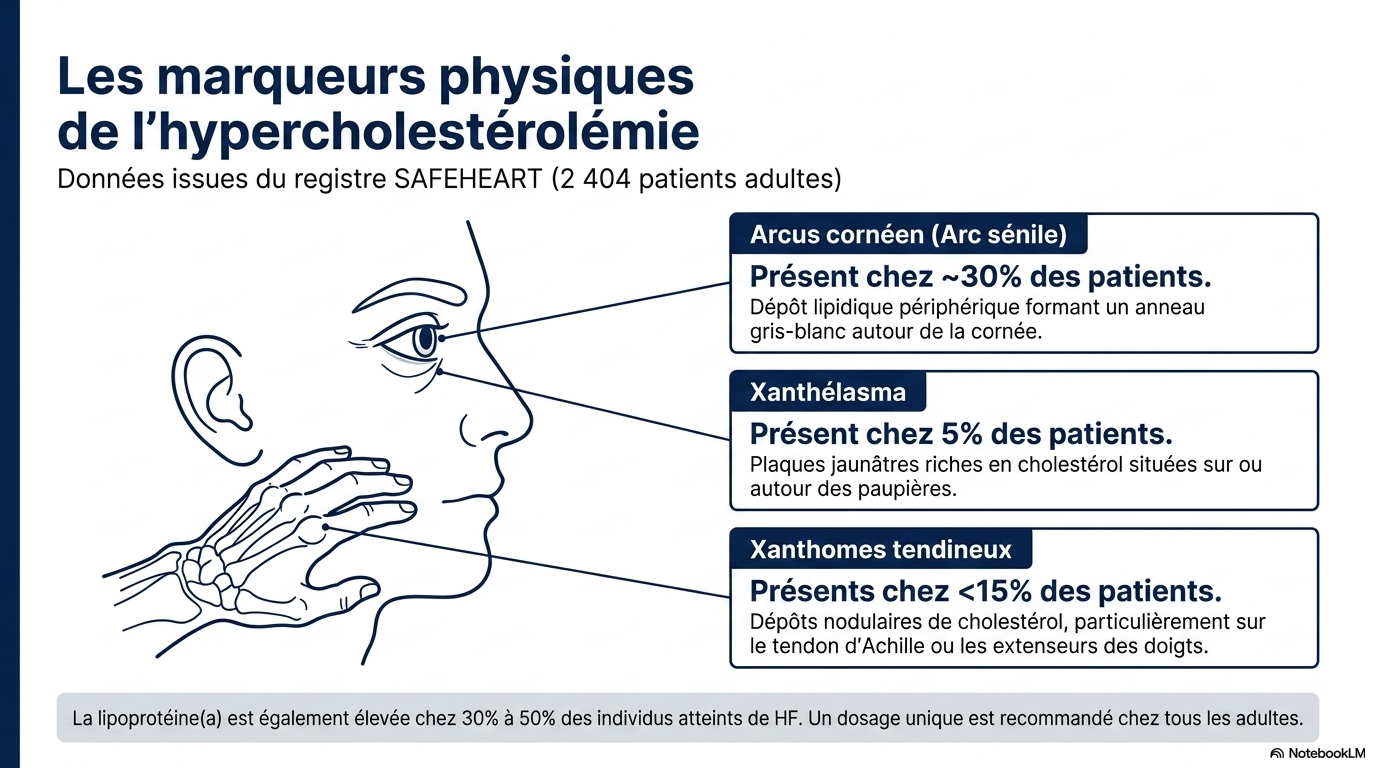
- xanthomes tendineux,
- arc cornéen précoce,
- xanthélasma.
Place du test génétique
Le séquençage des gènes LDLR, APOB et PCSK9 est recommandé chez les patients ayant une HF probable ou certaine.
Ses intérêts sont :
- confirmer le diagnostic ;
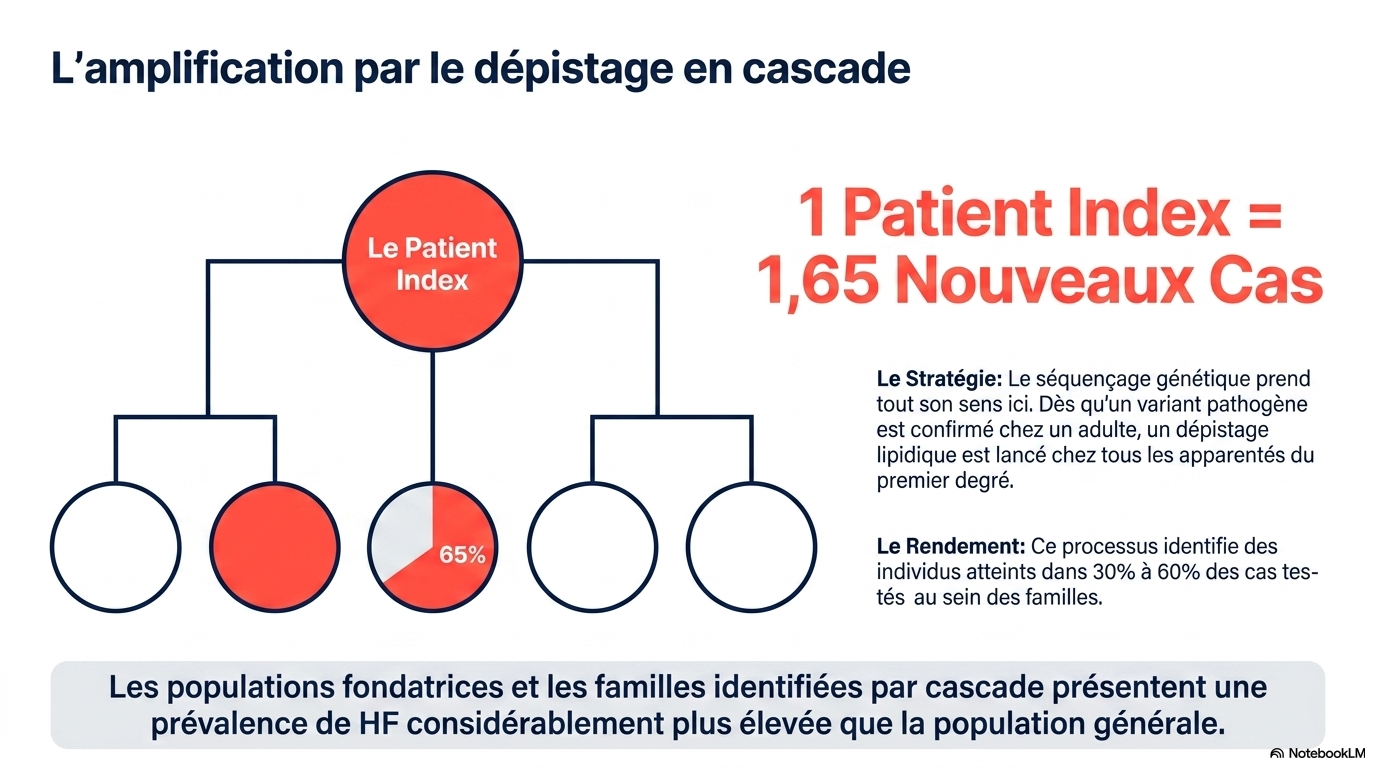
- faciliter le dépistage familial ("cascade screening") ;
- identifier les sujets les plus à risque ;
- faciliter l'accès à certaines thérapeutiques.
Cependant :
- seuls 2 à 3 % des adultes ayant un LDL ≥190 mg/dL présentent une mutation identifiable ;
- le traitement repose essentiellement sur le niveau de LDL et le risque cardiovasculaire, plus que sur le résultat génétique.
Dépistage
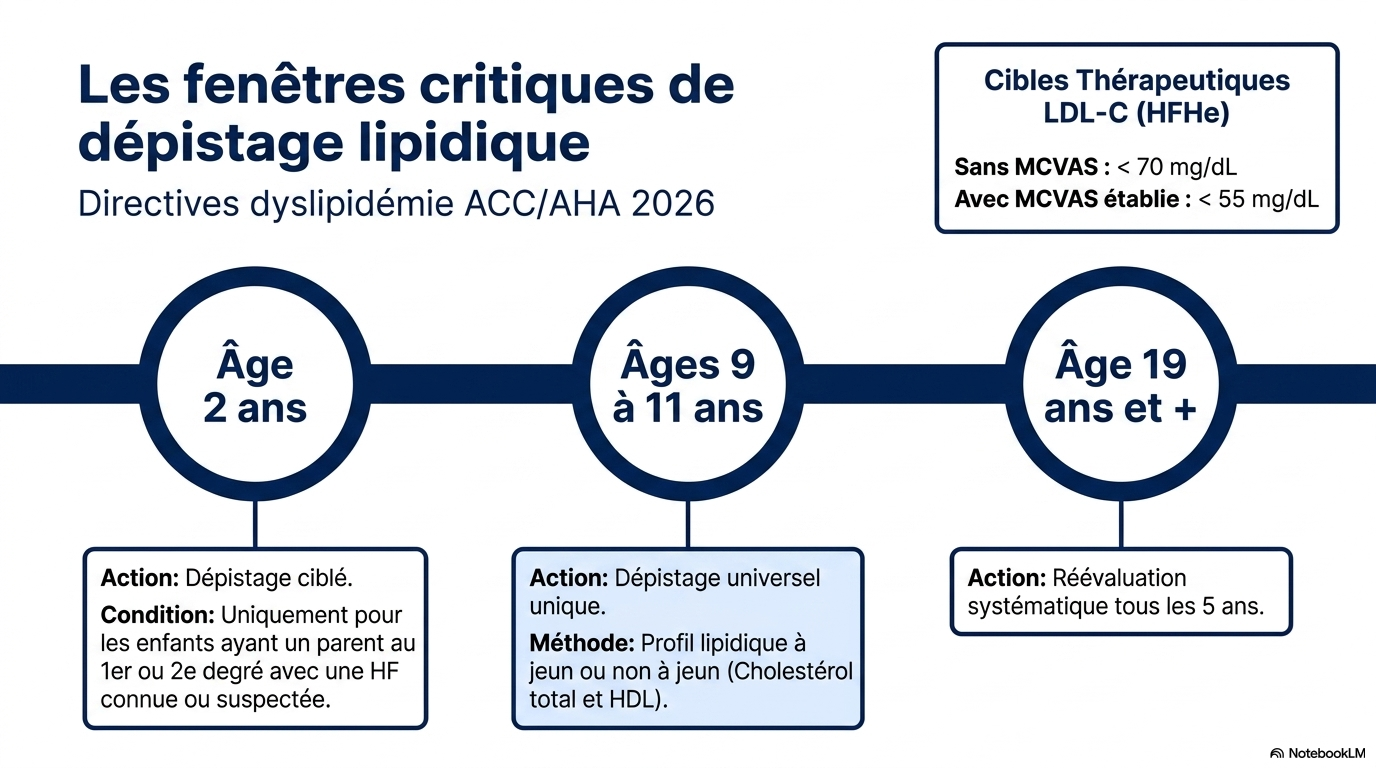
Les nouvelles recommandations ACC/AHA 2026 préconisent :
- dépistage lipidique universel entre 9 et 11 ans ;
- dépistage dès 2 ans chez les enfants ayant un apparenté atteint ;
- dosage unique de la Lp(a) chez tous les adultes.
Objectifs thérapeutiques
Les objectifs de LDL sont désormais très stricts :
- <70 mg/dL en prévention primaire (HF sans maladie cardiovasculaire).
- <55 mg/dL en prévention secondaire (HF avec ASCVD).
Traitement
Mesures hygiéno-diététiques
- diminution des graisses saturées (<7 % des calories) ;
- activité physique ≥150 minutes/semaine ;
- contrôle pondéral.
Traitement médicamenteux
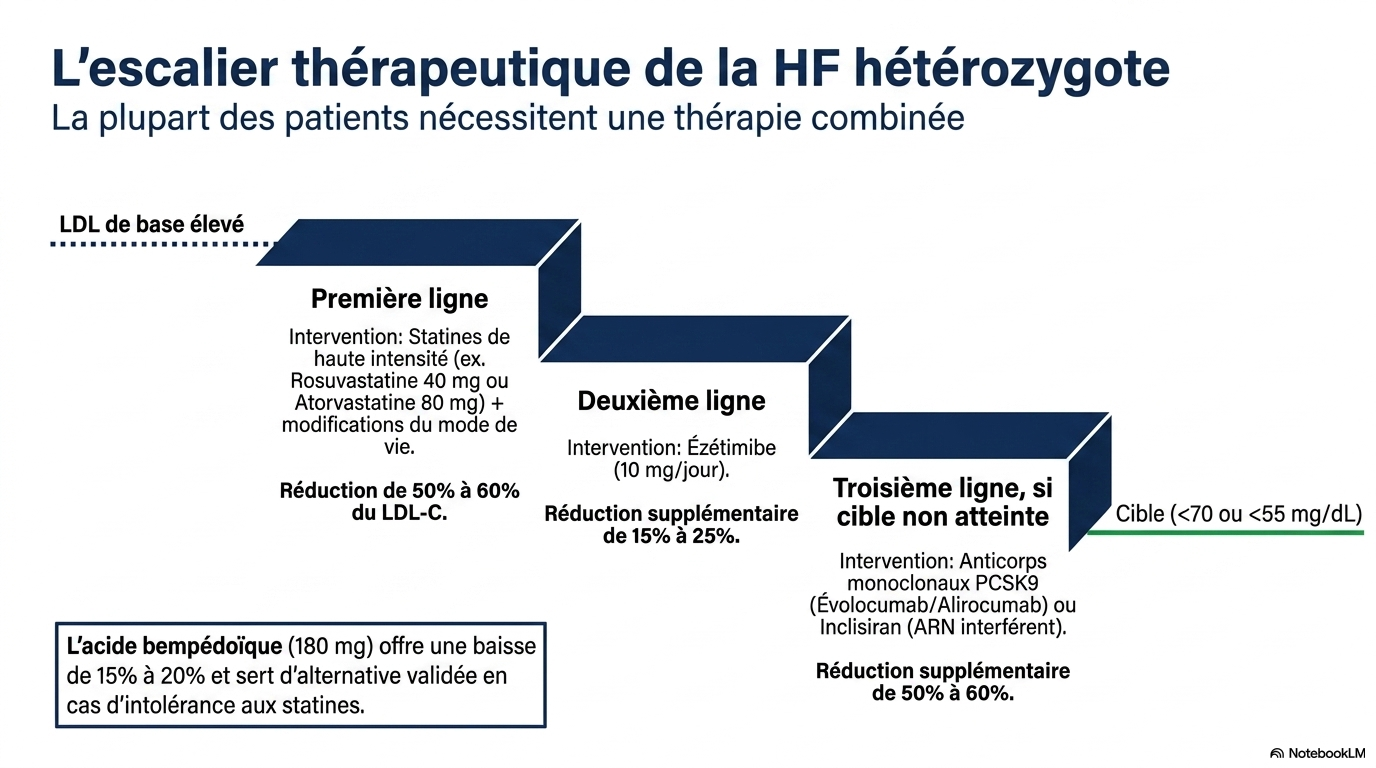
1. Statines fortes doses (1re intention)
- rosuvastatine 40 mg ;
- atorvastatine 80 mg.
➡ réduction du LDL de 50 à 60 %.
2. Ézétimibe
- réduction supplémentaire de 15 à 25 %.
3. Si objectif non atteint
- inhibiteurs de PCSK9 (évolocumab ou alirocumab) : –50 à –60 % supplémentaires ;
- inclisiran (2 injections/an) : efficacité comparable ;
- acide bempédoïque : –15 à –20 %, particulièrement utile en cas d'intolérance aux statines.
La majorité des patients nécessitent une bithérapie, voire une trithérapie.
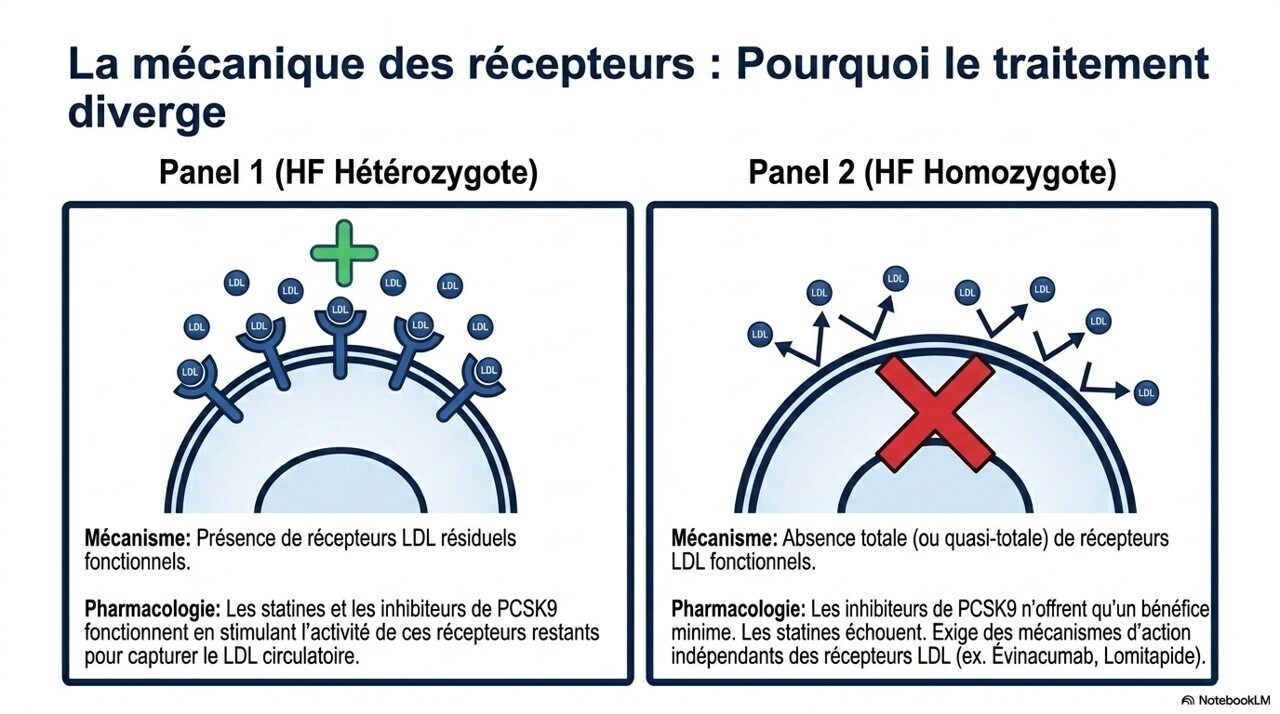
Hypercholestérolémie familiale homozygote
Cette forme sévère se caractérise par :
- LDL entre 400 et 1000 mg/dL ;
- diagnostic vers 12 ans en moyenne ;
- maladie cardiovasculaire souvent déjà présente.
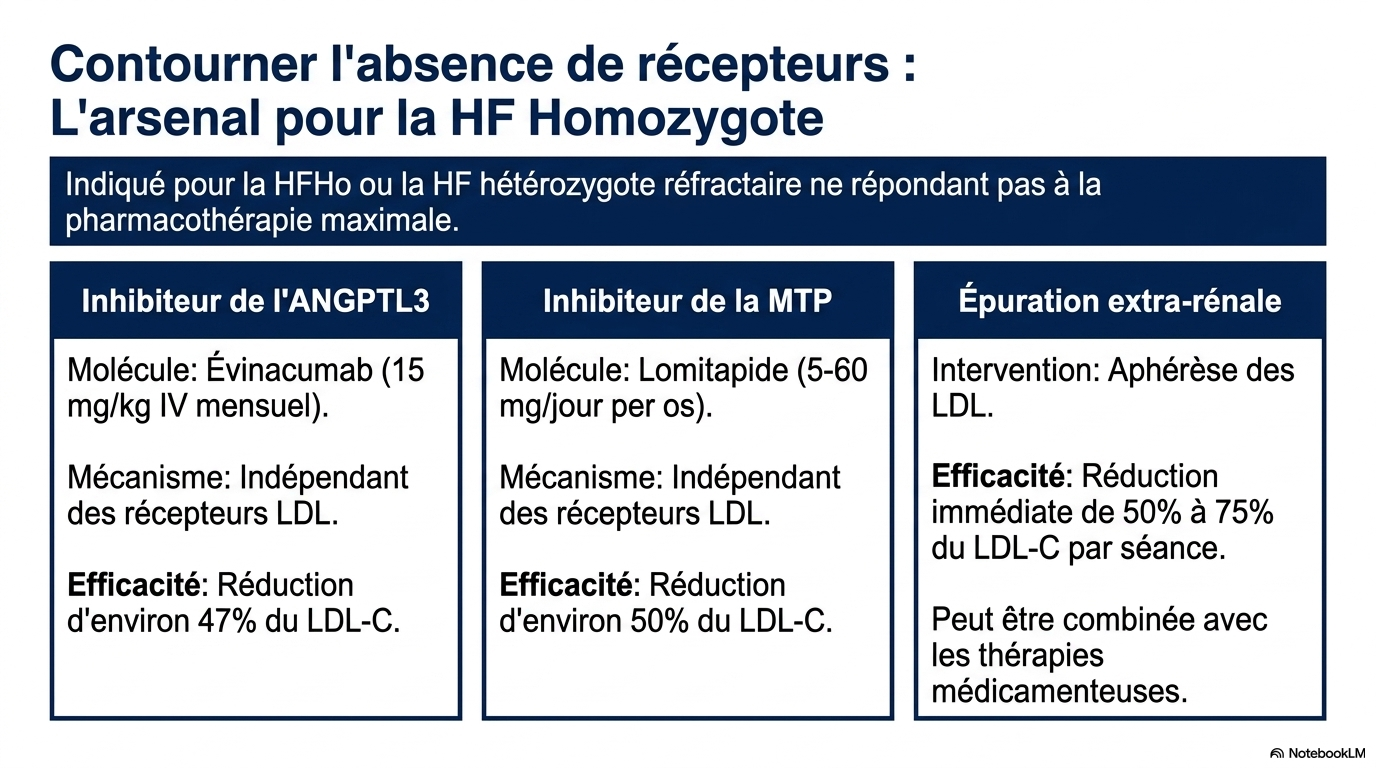
Les statines sont peu efficaces.
Les traitements spécifiques comprennent :
- évinacumab ;
- lomitapide ;
- aphérèse des LDL dans les formes sévères.
Les messages à retenir
- L'HF est fréquente mais largement sous-diagnostiquée.
- Un LDL ≥ 190 mg/dL doit faire rechercher une HF.
- Le diagnostic est essentiellement clinique.
- Le dépistage familial est indispensable.
- Plus le traitement est instauré tôt, meilleur est le bénéfice cardiovasculaire.
- Les objectifs thérapeutiques sont ambitieux (<70 ou <55 mg/dL selon le risque).
- La plupart des patients auront besoin d'une association de plusieurs hypolipémiants pour atteindre ces objectifs.
La prévalence de l'hypercholestérolémie familiale (HF) hétérozygote en France est estimée à environ 1 personne sur 120 (0,85 %) dans la population adulte de 35 à 74 ans, selon les données des enquêtes françaises MONICA et MONALISA utilisant les critères du Dutch Lipid Clinic Network (DLCN) modifiés. Ce chiffre est notablement plus élevé que les estimations mondiales poolées.
Données françaises spécifiques
L'étude de Bérard et al. (2019), basée sur 7 928 participants issus de 3 régions françaises (recrutés en 1995 et 2005), a retrouvé une prévalence d'HF « définie ou probable » de 0,85 % (IC 95 % : 0,63–1,06), soit environ 1 sur 120.
Parmi ces patients :
-
12 % avaient un antécédent de maladie cardiovasculaire prématurée.
-
70 % étaient traités (dont seulement 13 % par statine de haute intensité)
-
Aucun n'atteignait la cible de LDL-C recommandée, avec un écart moyen de 3,0 mmol/L par rapport à l'objectif
Comparaison avec les données mondiales
Les grandes méta-analyses internationales estiment la prévalence de l'HF hétérozygote dans la population générale à environ 1 sur 311–313. [2-3] Deux méta-analyses indépendantes portant sur plus de 7 et 11 millions de sujets convergent vers cette estimation.La prévalence est 10 à 20 fois plus élevée chez les patients atteints de maladie cardiovasculaire athéroscléreuse, atteignant environ 1 sur 16–17 chez les coronariens.
Sous-diagnostic et sous-traitement
L'HF reste largement sous-diagnostiquée en France comme dans le reste du monde : moins de 1 % des cas sont identifiés dans la plupart des pays. L'European Atherosclerosis Society souligne l'urgence d'un dépistage systématique et d'un traitement précoce et intensif. L'étude française de Fourgeaud et al. (2022) confirme que le diagnostic génétique d'HF monogénique peut être optimisé par le dépistage chez l'enfant (taux de LDL-C élevé), avec un dépistage en cascade inversé des apparentés.
L'écart entre la prévalence estimée en France (~1/120) et la prévalence mondiale (~1/311) peut s'expliquer en partie par les critères diagnostiques utilisés (DLCN clinique sans test génétique), la tranche d'âge étudiée (35–74 ans, excluant les sujets jeunes avec LDL-C encore modéré), et les caractéristiques populationnelles.
Commentaire
L'HYPERCHOLESTÉROLÉMIE FAMILIALE (HF) est sous-diagnostiquée et sous-traitée comme de nombreuses affections "étiquetées" rares.
Les marqueurs physiques cliniques de l'HF sont à rechercher :
ESC DYSLIPIDÉMIE 2025
- Risque très élevé/extrême : HF associée à une maladie cardiovasculaire athéroscléreuse (MCVA) documentée ou à un autre facteur de risque majeur.
- Risque élevé : HF sans autre facteur de risque majeur.
- Objectif : atteindre une cible de LDL-C < 0,70 g/L (1,8 mmol/L) ET une réduction minimale de 50 %
https://medvasc.info/archives-blog/dyslipidemies-esc-2025
