
Problèmes de prise en charge des malformations vasculaires
"La biotechnologie permettant désormais d'envisager des interventions conséquentes sur le génome humain, on assiste à la disparition progressive de la frontière entre la nature que nous sommes et l'équipement organique dont nous nous dotons." Jurgen Habermas
Analyse rédigée par :
"La connaissance du génome donnera de nouveaux outils pour comprendre comment l'individu s'adapte et se construit historiquement à partir de son génome sans que le génome constitue pour autant un destin. " Alain Prochiantz
Analyse rédigée par :

Pr Sophie Blaise
PU PH Medecine Vasculaire à l' Université Grenoble Alpes
CHU Grenoble
Présidente CFPV 2023
https://www.linkedin.com/in/sophie-blaise-45622511a/
https://www.linkedin.com/in/sophie-blaise-45622511a/
Merci Sophie d'avoir su nous emmener au coeur des malformations lymphatiques.
Montaser F ShaheenJulie Y TseEthan S SokolMargaret MastersonPranshu BansalIan RabinowitzChristy A TarletonAndrey S DobroffTracey L SmithThèrése J BocklageBrian K MannakeeRyan N GutenkunstJoyce BischoffScott A NessGregory M RiedlingerRoman GroisbergRenata PasqualiniShridar GanesanWadih Arap (2022) Genomic landscape of lymphatic malformations: a case series and response to the PI3Kα inhibitor alpel
Paysage génomique des malformations lymphatiques : une série de cas et une réponse à l'alpelisib, un inhibiteur de PI3Kα
Article libre d'accès
https://elifesciences.org/articles/74510
Les malformations vasculaires posent régulièrement des problèmes de prise en charge. Les lymphatiques (ML) posent souvent des problèmes de traitement en raison d'une grande taille ou d'uneune localisation particulièrement problématique, notamment car elles sont souvent localisées au niveau de la face.
Pour celles de large volume et non chirurgicale, le traitement de référence est la sclérothérapie, mais la conduite à tenir thérapeutique n’est pas standardisée.
Depuis quelques années, les approches génomiques ont un peu changé la donne. Il est présenté dans cet article une cohorte de 30 cas de malformations lymphatiques avecleurs analyses génomiques et l’illustration de l’efficacité de l’alpelisib chez un de ces patients non répondeur à la rapamycine.
Pour celles de large volume et non chirurgicale, le traitement de référence est la sclérothérapie, mais la conduite à tenir thérapeutique n’est pas standardisée.
Depuis quelques années, les approches génomiques ont un peu changé la donne. Il est présenté dans cet article une cohorte de 30 cas de malformations lymphatiques avecleurs analyses génomiques et l’illustration de l’efficacité de l’alpelisib chez un de ces patients non répondeur à la rapamycine.
Méthodes
Etude d’une cohorte de 30 patients avec ML ayant subi un profilage génomique complet à l'aide d'un test de séquençage de nouvelle génération à large panel ainsi que des analyses immunohistochimiques.
Etude d’une cohorte de 30 patients avec ML ayant subi un profilage génomique complet à l'aide d'un test de séquençage de nouvelle génération à large panel ainsi que des analyses immunohistochimiques.
Résultats
Les patients étaient principalement d'âge pédiatrique (médiane de 9 ans; intervalle de 1 à 45 ans), avec une légère prédominance féminine (17 femmes, 57 % – 13 hommes, 43 %). Sept patients avaient un antécédent de traitement antérieur par un inhibiteur de mTOR, tel que le sirolimus. Sept patients (23 %) avaient un diagnostic clinique de type syndrome de prolifération, de type CLOVES, Klippel–Trenaunay ou PTEN.
Douze patients (40 %) avaient une maladie multifocale et huit patients avaient une atteinte osseuse et sur des sites viscéraux.
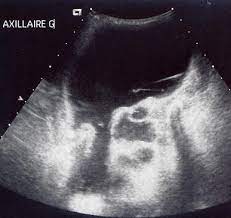 Malformation lymphatique fcreux axillaire, adolescente, Collection JPL/M Dauzat
Malformation lymphatique fcreux axillaire, adolescente, Collection JPL/M Dauzat
Les patients étaient principalement d'âge pédiatrique (médiane de 9 ans; intervalle de 1 à 45 ans), avec une légère prédominance féminine (17 femmes, 57 % – 13 hommes, 43 %). Sept patients avaient un antécédent de traitement antérieur par un inhibiteur de mTOR, tel que le sirolimus. Sept patients (23 %) avaient un diagnostic clinique de type syndrome de prolifération, de type CLOVES, Klippel–Trenaunay ou PTEN.
Douze patients (40 %) avaient une maladie multifocale et huit patients avaient une atteinte osseuse et sur des sites viscéraux.
Malformation lymphatique fcreux axillaire, adolescente, Collection JPL/M DauzatLe profilage mutationnel a montré que ces ML avaient une charge mutationnelle tumorale uniformément faible.
Les mutations PIK3CA (n = 20) et les mutations NRAS (n = 5) étaient les plus fréquentes et s'excluaient mutuellement.
Tous les cas de ML avec une histologie de type sarcome de Kaposi (kaposiforme) avaient des mutations NRAS. Les fréquences d'allèles variants (VAF) des mutations PIK3CA et NRAS étaient relativement faibles (médiane, 6 %; intervalle, 1 à 38 %), compatible avec un pourcentage estimé histopathologique relativement faible de noyaux tumoraux. Ces résultats suggèrent que les mutations PIK3CA et NRAS étaient probablement clonales.
Tous les cas de ML avec une histologie de type sarcome de Kaposi (kaposiforme) avaient des mutations NRAS. Les fréquences d'allèles variants (VAF) des mutations PIK3CA et NRAS étaient relativement faibles (médiane, 6 %; intervalle, 1 à 38 %), compatible avec un pourcentage estimé histopathologique relativement faible de noyaux tumoraux. Ces résultats suggèrent que les mutations PIK3CA et NRAS étaient probablement clonales.
L'analyse histopathologique des lésions a identifié que 4 des spécimens analysés présentaient des caractéristiques histopathologiques kaposiformes avec une prolifération cellulaire. Les 26 lésions restantes (87 %) avaient des histopathologies conventionnelles. caractéristiques typiques d’une ML classique, avec prolifération de vaisseaux lymphatiques dilatés à parois minces avec ou sans matière protéique luminale. Parmi les cas de ML d'histologie conventionnelle, 20 (77 %) avaient un mutation PIK3CA, tandis que 1 (4 %) avait une mutation NRAS, et 5 (19%) étaient de type sauvage pour les deux gènes, dont un seul cas avec une fusion génétique GOPC-ROS1. Les 4 cas de LM avec kaposi (KLM) avaient une mutation NRAS activatrice, compatible avec l'enrichissement de la mutation NRAS (p =
0,00018) et absence de mutation PIK3CA dans cette histologie (p = 0,0046).
Un patient s'est présenté avec des douleurs abdominales et a été diagnostiqué avec une ML rétropéritonéale avec une mutation somatique de gain de fonction PIK3CA . Le patient a obtenu une réponse radiologique complète rapide et durable, à l'alpelisib, un inhibiteur de la PI3Kα, dans le cadre d'un essai clinique personnalisé N-sur-1 (NCT03941782), qui à l’époque était encore expérimental (non approuvé par FDA). Le patient a été mis sous alpelisib quotidiennement dose de 350 mg par voie orale et il a signalé une régression de son renflement abdominal en quelques jours. Aucun effet indésirable n’a été mentionné (pas de variation glycémique). Un scanner abdominal à 6 semaines montrait une régression de la ML. La réponse était complète un an après le début de l’essai. Après 2 ans, l’alpelisib a été
interrompu en raison de problèmes théoriques sur l'impact négatif à long terme sur l’homéostasie. Malheureusement, la masse a récidivé au bout de quelques semaines et le patient a donc repris l’alpelisib avec une deuxième réponse partielle maintenue depuis plus de 3 ans
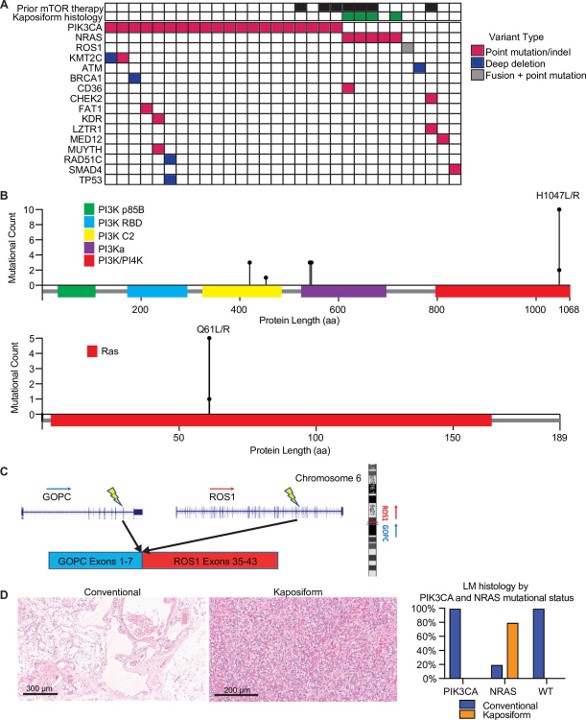
Paysage mutationnel et histopathologie des malformations lymphatiques (LM).
( A ) Oncoprint montrant le paysage mutationnel de 30 échantillons LM séquencés. ( B ) Parcelle Lollipop montrant le spectre des mutations PIK3CA et NRAS dans cette cohorte. ( C ) Schéma montrant les détails de la fusion GOPC – ROS1 identifiée dans un LM de type sauvage NRAS et PIK3CA . ( D ) Images histologiques représentatives pour les LM avec histologie conventionnelle et kaposiforme. Les fréquences relatives des mutations PIK3CA et NRAS dans les deux variantes histologiques sont tracées.
Paysage mutationnel et histopathologie des malformations lymphatiques (LM).
( A ) Oncoprint montrant le paysage mutationnel de 30 échantillons LM séquencés. ( B ) Parcelle Lollipop montrant le spectre des mutations PIK3CA et NRAS dans cette cohorte. ( C ) Schéma montrant les détails de la fusion GOPC – ROS1 identifiée dans un LM de type sauvage NRAS et PIK3CA . ( D ) Images histologiques représentatives pour les LM avec histologie conventionnelle et kaposiforme. Les fréquences relatives des mutations PIK3CA et NRAS dans les deux variantes histologiques sont tracées.
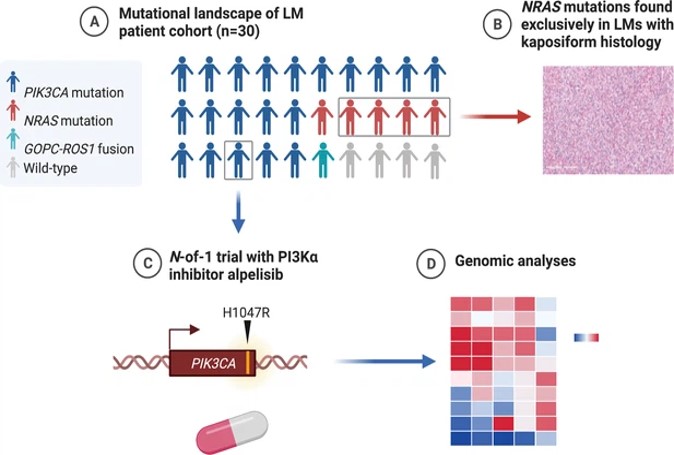
Résumé graphique des mutations trouvées dans l'analyse génomique de la cohorte de patients atteints de malformation lymphatique (LM) (créée avec BioRender.com).( A ) La majorité des LM ont des mutations motrices qui sont potentiellement ciblables. ( B ) Les LM avec des mutations NRAS avaient une histopathologie kaposiforme. ( C ) Un essai clinique N -sur-1 est rapporté chez un patient présentant une mutation PIK3CA ciblable. ( D ) Des analyses génomiques complètes peuvent révéler d'autres informations moléculaires exploitables.
Discussion
Dans des études de corrélation translationnelle, l'activation de la voie PI3Kα a été confirmée par immunohistochimie et les cellules endothéliales lymphatiques humaines dérivées de ML portant un allèle avec une mutation activatrice au même locus étaient sensibles au traitement par l'alpelisib in vitro. Ce cas clinique confirme cette piste translationnelle.
Dans des études de corrélation translationnelle, l'activation de la voie PI3Kα a été confirmée par immunohistochimie et les cellules endothéliales lymphatiques humaines dérivées de ML portant un allèle avec une mutation activatrice au même locus étaient sensibles au traitement par l'alpelisib in vitro. Ce cas clinique confirme cette piste translationnelle.
Les KLM ont des caractéristiques cliniques, histologiques et génomiques distinctes.Cliniquement, ils sont plus susceptibles de survenir chez les patients jeunes et sous une formegénéralisée avec atteinte du médiastin, plèvre et péricarde. Histologiquement, ils sontcomposés de feuillets très cellulaires et nodulaires, proliférations de cellules fusiformes,rappelant le sarcome de Kaposi. Contrairement au sarcome de Kaposi, les cellules tumorales ne sont pas positives à l' herpès virus humain-8. Sur le plan génomique, des études récentes ont montré qu'ils ont tendance à être mutée NRAS. Un biais est soulevé de l’absence de mutation parfois du fait d’un prélèvement insuffisant. Il est important de noter que 3 sur 5
patients atteints de NRAS- avaient échoué au traitement par sirolimus. Certains ML avec NRAS pourraient répondre au traitement avec des inhibiteurs de MEK, suggérant que cela peut être une option pour les ML avec des caractéristiques kaposiformes. Ces données suggèrent que la plupart des ML peuvent avoir des mutations PIK3CA dominant (ML avec une histologie conventionnelle) et des mutations NRAS (ML avec des caractéristiques kaposiformes).
Faits intéressants, chez les malades avec ML sans tissu solide disponible pour des prélèvements avec séquençage génomique, une biopsie liquide pour un séquençage réalisé sur une tumeur circulante ADN dans le sang périphérique pourrait être une solution possible pour les ML. Elles sont associées de manière innée avec le système vasculaire et donc potentiellement avec excrétion d'ADN dans le sang périphérique.
Le cas clinique suggère que les inhibiteurs de PI3Kα n'éliminent pas complètement tous cellules initiatrices de ML, et qu’il peut être nécessaire de les administrer à long terme pour un contrôle durable.
Cette classe de médicaments pourrait être envisagée dans une approche néoadjuvante pour rendre certaines formes résécables. Attention, des mécanismes de résistance acquise aux inhibiteurs de PI3Kα ont été rapportés, en raison d'autres mutations compensatrices ou de contournement associées. telles que celles impliquant l'oncogène RAS ou le gène suppresseur de tumeur PTEN, et ceux-ci peuvent éventuellement survenir chez ces patients avec un suivi plus long dans le temps.
Perspectives
Depuis cet article, la FDA a approuvé l'alpelisib pour les patients pédiatriques et adultes atteints de PROS, et plusieurs essais cliniques sont en cours.
Les preuves s'accumulent qu'une variété de syndromes « non malins » associés à de la croissance tissulaire peut être induite par des altérations sous-jacentes des oncogènes classiques. Les mutations de PIK3CA sont observées non seulement dans les ML mais aussi dans d'autres anomalies vasculaires, mettant en évidence le rôle de l'activation de PIK3CA dans l'angiogenèse, la lymphangiogenèse et les néoplasmes vasculaires. Les thérapies ciblées développées pour les cancers invasifs pourraient également être actives dans les lésions prolifératives qui ne sont pas classées comme cancers invasifs en cas de mutation ciblée
Depuis cet article, la FDA a approuvé l'alpelisib pour les patients pédiatriques et adultes atteints de PROS, et plusieurs essais cliniques sont en cours.
Les preuves s'accumulent qu'une variété de syndromes « non malins » associés à de la croissance tissulaire peut être induite par des altérations sous-jacentes des oncogènes classiques. Les mutations de PIK3CA sont observées non seulement dans les ML mais aussi dans d'autres anomalies vasculaires, mettant en évidence le rôle de l'activation de PIK3CA dans l'angiogenèse, la lymphangiogenèse et les néoplasmes vasculaires. Les thérapies ciblées développées pour les cancers invasifs pourraient également être actives dans les lésions prolifératives qui ne sont pas classées comme cancers invasifs en cas de mutation ciblée
Conclusion
La majorité des LM ont des mutations qui sont potentiellement ciblables (PIK3CA et NRAS) et peuvent avoir d'autres modifications exploitables, telles que la fusion GOPC-ROS1. Un traitement systémique avec une thérapie ciblée visant la mutation peut être une option pour certains patients qui ne sont pas contrôlés par la chirurgie et d'autres traitements conventionnels. Les prélèvements pourraient être réalisés sur biopsie de liquide ou sanguin en cas d’absence de possibilité de prélèvement sur tissu.
La majorité des LM ont des mutations qui sont potentiellement ciblables (PIK3CA et NRAS) et peuvent avoir d'autres modifications exploitables, telles que la fusion GOPC-ROS1. Un traitement systémique avec une thérapie ciblée visant la mutation peut être une option pour certains patients qui ne sont pas contrôlés par la chirurgie et d'autres traitements conventionnels. Les prélèvements pourraient être réalisés sur biopsie de liquide ou sanguin en cas d’absence de possibilité de prélèvement sur tissu.
Fiche médecin alpelisib

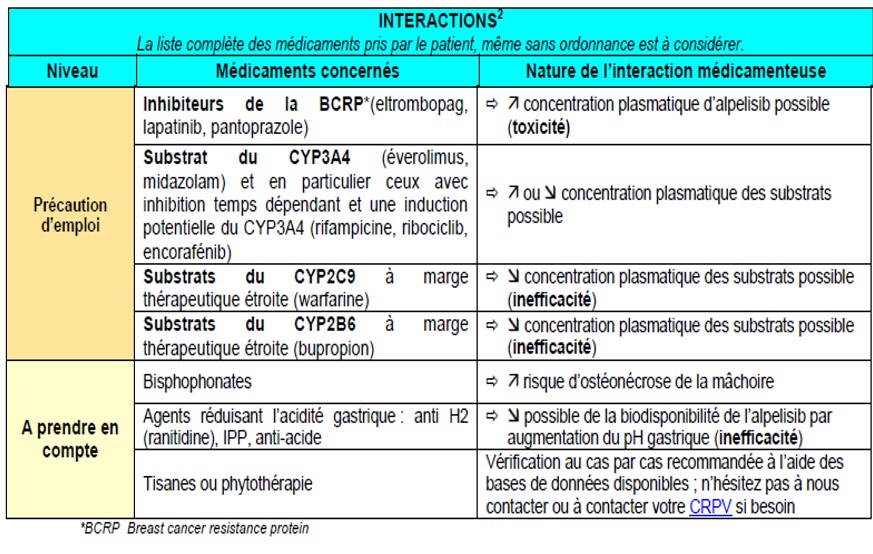
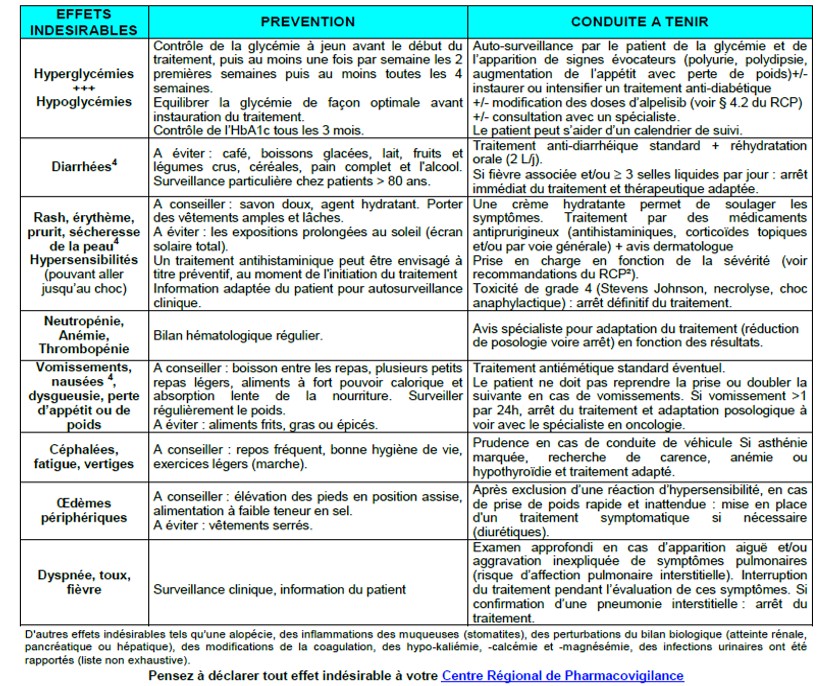

https://www.omedit-paysdelaloire.fr/wp-content/uploads/2020/06/alpelisib-v1_0-pro.pdf