iconographie : VEXAS
Un grand merci d'avoir accepté.

Hématologiste
Service d'Hématologie biologique de l'Hôpital européen Georges Pompidou avec le Pr D SMADJA (cheffe de service, Pascale GAUSSEM)
Université de Paris, Innovation Thérapeutique en Hémostase, INSERM UMRS 1140 et Laboratoire de Recherches Biochirurgicales (Fondation Carpentier), Paris, France
Service d'hématologie biologique, Hôpital européen Georges Pompidou, Paris, France
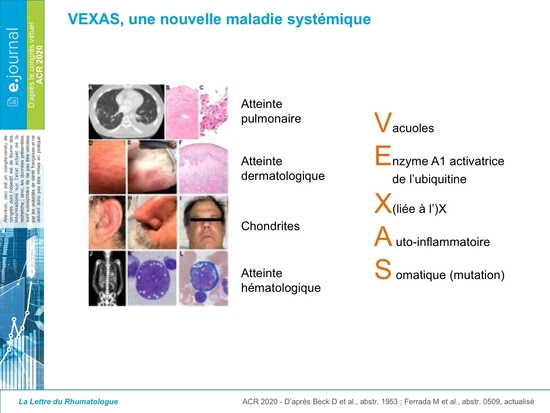
Syndrome VEXAS . qu'est-ce que c'est ?
Le syndrome VEXAS (acronyme pour Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic) est une maladie auto-inflammatoire décrite chez l’adulte, causé par une mutation somatique du gène Ubiquitin like modifier activating enzyme 1 (UBA1) caractérisée en fin d’année 2020 aux états Unis et rapidement confirmée par plusieurs équipes Française.
Le syndrome VEXAS est majoritairement décrit chez les hommes avec une apparition progressive de la maladie après 50 ans. Les 25 patients de la cohorte initiale sont exclusivement des hommes, il a donc été suggéré que l’allèle du second chromosome X de la femme protégeait de l’action néfaste de l’allèle UBA1 muté. La description plus récente de plusieurs femmes VEXAS présentant au caryotype une monosomie du X conforte cette hypothèse. La vraie prévalence de ce syndrome est à ce jour inconnue mais les nombreuses descriptions tendent à monter que cette entité n’est pas si rare avec cependant une grande majorité de cas masculins atteints.
Typiquement les patients VEXAS présentent une maladie inflammatoire systémique avec des épisodes de fièvre inexpliqués, une atteinte pulmonaire, cutanée, vasculaire et articulaire. La majorité des patients répondent aux critères de diagnostic établis pour différentes maladies inflammatoires telles que le syndrome de Sweet, la polyartérite noueuse, la polychondrite récidivante. D’un point de vue hématologique, les patients VEXAS présentent des cytopénies et notamment une anémie macrocytaire (96%), un syndrome myélodysplasique (SMD, 24%), un myélome multiple (20%) et des évènement thromboemboliques.
Il s’agit d’une pathologie sévère, progressive et relativement résistante aux traitements jusqu’alors mis en place. Le taux de mortalité est élevé, de 25 % à 35 % à 5 ans.
Les complications thrombotiques sont rapportées chez environ 40% des patients VEXAS, avec une prédominance d’évènement thromboemboliques veineux (36.4%) plutôt qu’artériels (1.6%). A noter que la fréquence élevée d’
Le diagnostic est génétique doit être suspecté face à une maladie inflammatoire systémique ne répondant pas au traitement habituel ou face à un SMD associé à des signes inflammatoires systémiques,. Ce diagnostic moléculaire du syndrome VEXAS se fait par séquençage du gène UBA1. Il ne s’agit pas d’une analyse de routine, actuellement disponible en centres très spécialisés (par exemple au laboratoire d’Hématologie de l’Hôpital Cochin, Pr O Kosmider)
Chez qui le rechercher en cas de MTEV chez un homme de plus de 50 ans sans facteur déclenchant ?
Pour rappel, selon les dernières recommandations de bonne prise en charge de la MTEV (Sanchez O, Rev Mal Res 2019).
R132 -Chez les patients présentant un premier épisode non provoqué de MVTE, il est recommandé :
- d’effectuer un examen physique attentif et de recueillir les antécédents néoplasiques personnels et familiaux et de répéter cette évaluation au cours des six premiers mois de suivi et d’orienter les investigations en fonction des éventuelles anomalies observées (grade 1+) ;
- en dehors de la surveillance du traitement anticoagulant classique (ionogramme sanguin avec créatininémie, tests hépatiques), de réaliser une radiographie de thorax (si un scanner thoracique n’a pas été réalisé pour le diagnostic d’EP), une numération formule sanguine, calcémie (grade1+) ;
- de mettre à jour les dépistages recommandés dans la population générale (grade 1+) : réaliser un frottis chez toutes les femmes, une mammographie après50 ans, et un PSA chez tous les hommes de plus de50 ans, sauf si ces examens ont été réalisés dans l’année précédente ;
- les éventuelles autres explorations seront guidées par les résultats des premiers examens (grade 1+).
R138 — Il est recommandé de ne pas réaliser de bilan de thrombophilie constitutionnelle chez les patients ayant un premier épisode de TVP proximale ou d’EP après 50 ans que la thrombose soit provoquée ou non
Dans notre étude qui incluait 97 patients de sexe masculin issus de la cohorte FARIVE (Pr J Emmerich) inclus à l’HEGP entre 2003 et 2009 pour lesquels de l’ADN était disponible et ayant présenté un évènement thrombotique veineux (dont 50.5% d’évènement non provoqué) : aucun patient ne présentait cette mutation.
Ces résultats ne sont pas en faveur de la recherche de mutations de l'exon 3 d'UBA1 chez tous les hommes de plus de 50 ans ayant un premier événement thromboembolique veineux, sans tenir compte des autres symptômes cliniques pouvant être associés au VEXAS.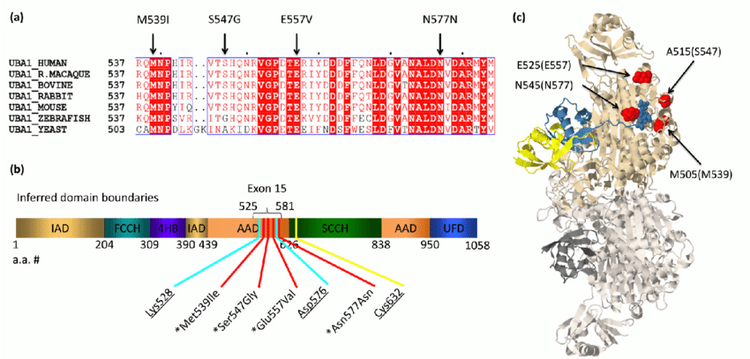 Mutation gène UBA1
Mutation gène UBA1
La présence de la mutation du gène UBA1 a quelles conséquences ?
Il s’agit d’une mutation somatique du gène Ubiquitin like modifier activating enzyme 1 (UBA1). Ce gène est situé sur le chromosome X et est impliqué dans le processus d’ubiquitination des protéines.
Chez les patients VEXAS, il est supposé que le défaut d’ubiquitination entraine une dérégulation de l’immunité innée et une inflammation systémique vasculaire favorisant la survenue de thrombose. En effet il est décrit une activation aberrante des monocytes et neutrophiles, un relargage important de cytokines qui participent à un état d’hypercoagulabilité. De plus, les vascularites rapportées chez certains de ces patients favorisent une dysfonction endothéliale participant au phénomène hypercoagulant. Il est à noter que les patients VEXAS présentent une formation accrue de NETS et que 44% des patients présentent des anticorps antiphospholipides persistants.
Merci Nicolas de cette mise au point qui était nécessaire et très utile.